



Genome Annotation Technology
Annotation by Protein Families
Annotation by Sub-Systems - Knowing What to Look For
Finding Genes for Functions - Positional Clustering
Protein Fusions - Guilt by Association
Occurrence Profiles
Other Guilt by Association Techniques
Towards 100% Reconstruction
Traditional genome annotation includes finding genes via statistical methods, assigning functions based on similarities, and then manual gene-by-gene analysis. During this process, only 40-50% of the found genes are assigned a function, with average 5-15% false assignment rate, while core metabolic machinery usually gets covered by up to 60%.
Genome Designs practices an alternative method of annotation, which uses other types of inference in addition to structural similarity of proteins:
- Conservatism in protein structure-to-function relationships (each function is associated with a limited number of structures)
- Evolutionary conservatism of functional designs (core machinery and peripheral subsystems)
- Tendency of functionally related genes to physically cluster in genomes
- Tendency of functionally related proteins to fuse
- Co-regulation of functionally related genes
- Mutual dependencies in expression of functionally related genes
- Likelihood of protein-protein interactions between functional counterparts.
Most of the existing genome analysis systems offer tools to retrieve the above information, but rely upon annotators to actually assign functions. Human-made annotations are propagated through the traditional annotation mechanism to similar proteins, amplifying possible mistakes.
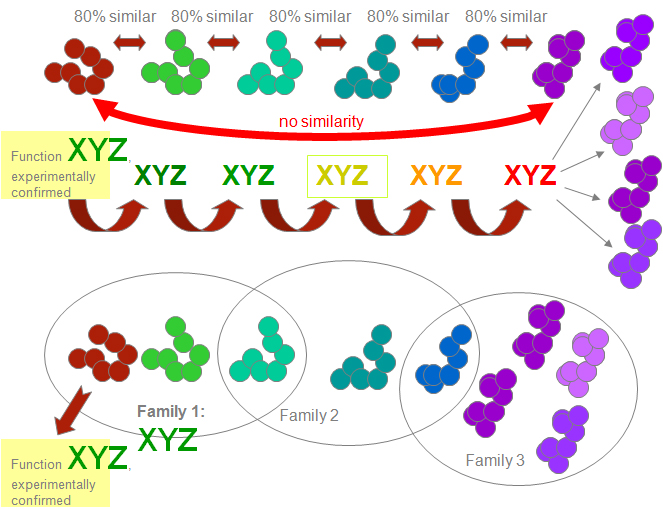
Annotation by Protein Families
Proteins are traditionally assigned functions from structurally similar proteins with known functions. As the process repeats from one similar protein to another, similarity to an original protein may fade. Thus, a protein can be assigned a wrong function, and this wrong assignment can get propagated to other similar proteins.
To avoid this pitfall, we are relying on the fact that proteins are classified into families in which all members are structurally related to one another. A family in turn can be associated with a function if there is good evidence.
Genome Designs applies the same technique to other types of inference: we try to interpret clusters, not individual elements.
Annotation by Sub-Systems - Knowing What to Look For
Core Functional Machinery of Life is a highly conserved network of metabolic and information-processing functions (about 2000 distinct functional roles). It can be viewed as a set of distinct functional sub-systems (often referred as pathways), each providing for a certain functionality.
- Modular design: every sub-system appears to have a very limited number of implementations (usually just one).
- Functional completeness principle: the entire subsystem either exists or not in any particular organism. The appearance of one of its functions infers the appearance of the entire pathway.
- Subsystems always compose a logically complete design and follow certain standard design commitments: some are obligatory (for example, every organism must replicate DNA or produce ATP); others depend on each other (if there is a Citric Acid cycle, there must be a way to synthesize Acetyl-CoA).
Peripheral subsystems are less ubiquitous but also highly standardized: transport; secondary metabolism; motility; etc.
Traditional approach implies finding function for a gene, where search space is large and unstructured. Our method includes finding gene for a function, with a number of ways to rationalize the search by evolutionary inference. It allows to concentrate on the practically important genes, and helps finding genes even if the corresponding ORFs are not called.
Finding Genes for Functions - Positional Clustering
Functionally related genes tend to form clusters on chromosomes in bacteria and archaea:
- Horizontal gene transfer preserves functionally complete cassettes, giving a competitive advantage to the recipient
- Adjacent genes participate in operons and provide better regulation options
- 90% of Core Machinery pathways can be found by blind analysis of chromosomal clustering in bacteria
- Clustering in higher organisms is less expressed, but also observable, especially in Fungi
Location of individual genes is mostly random. Clustering applies to the families of structurally related proteins. If members of a certain protein family appear on chromosomes in close proximity to members of another protein family at a frequency higher than random, then proteins of these families are likely to play related functional roles.
Protein Fusions - Guilt by Association
If members of a certain protein family appear as domains of fused proteins together with members of another protein family, the proteins of these families are likely to play related functional roles. Fusions of unrelated proteins occur with a much lower frequency.
Occurrence Profiles
Why do members of protein families that are associated with related functional roles tend to appear in organisms together? The functional completeness principle implies that organisms have complete pathways. At the same time, the structural conservatism principle requires each functional role to be associated with only a limited number of protein families. As the result, we can expect similar combinations of protein families in all organisms that carry a particular pathway.
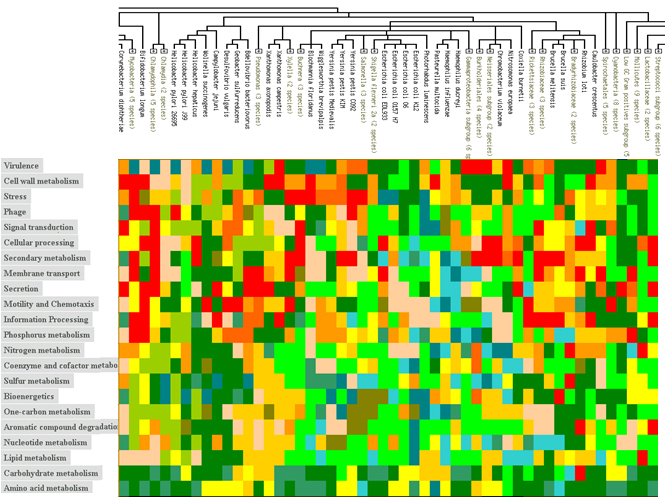
Other Guilt by Association Techniques
Participation in co-regulation suggests a functional relationship. Co-expression (or more complex dependency of expression profiles) suggests a functional relationship. These effects become significant only when observed on many members of a protein family. This way, the annotation for a member of a protein family infers from the properties of the family, not from those of its own.
Towards 100% Reconstruction
Efficiency of an average reconstruction of bacteria):
- Similarity-based assignment (auto):40-50%
- Orthology (COG-based): adds 5-15%
- With the use of Subsystems and Pathways:
- Chromosomal clustering: adds 15-25%
- Fusion events: adds 10-15%
- Co-occurrence: adds 10-15%
- Co-regulation: up to 10% (estimated)
- Co-expression: up to 15% (estimated)
- Domain families and motifs: up to 10% (estimated)
Total effect: up to 70-80% proteins get functions. where up to 95% belong to functional families (15-25% of functions are unknown)
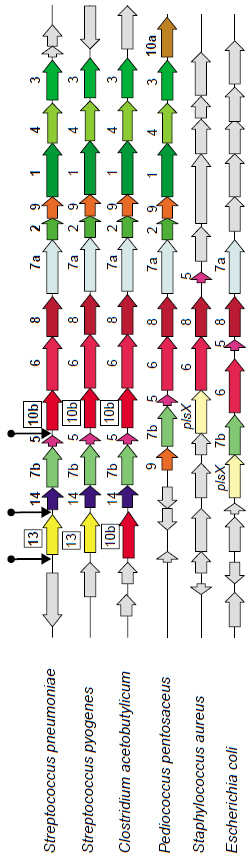
A gene arrangement in P. pentosaceus is very similar to S. pneumoniase, S. pyogenes and C. acetobutylicum, with a most notable disappearance of fabK (10b) in the middle of the cluster compensated by the appearance of fabI (10a) at the end of the cluster.
From: Missing genes in metabolic pathways: a comparative genomics approach by A. Osterman and R. Overbeek,
Current Opinion in Chemical Biology 2003